.
Introduction
Autism spectrum disorders are characterized by a behavioral syndrome that is recognized between ages 2 and 3 years. The core of the syndrome is a deviant and (or) retarded development of cognitive capacities and skills necessary for social relations, communication, fantasy, and symbolic thinking. Almost all autistic people do not reach independence as adults, and 75% are mentally retarded as well (Shaw, Kassen, and Chaves, 1995). Despite the fact that autism is considered by many leading researchers to be one of the most genetic of all multifactorial neurodevelopmental disorders, with a concordance rate of over 50% between monozygotic (identical) twins versus less than 5% for dizygotic (fraternal) twins (Previc, 2007) and despite the fact that those with autism rarely marry and thereby pass on their genes, there has been a dramatic increase in autism in all industrialized nations (and especially the United States) over the past 25 years which is difficult to explain based solely based on genetic factors. Even in the case of the recently discovered ARHGEF6 gene single nucleotide polymorphism (SNP) on the X-chromosome, which accounts for about 30% of the cases of autism in males, 20% of those males with this gene SNP do not have autism (Pratt-Hyatt, Haeusler, Yuen, and Shaw, 2018).
Abnormalities in the metabolism of dopamine in autism have been reported in the research literature for 40 years (Lelord et al (1978). Both Lelord et al (1978) and Garreau et al (1980) reported that urinary HVA levels were higher in autistic than in normal children. LeLord et al (1978) reported that the clinical improvement of these young autistic patients under vitamin B6 treatment was associated with a decrease of their urinary HVA excretion. Abnormalities in dopamine metabolism include elevated concentrations of the dopamine metabolite homovanillic acid (HVA) in cerebrospinal fluid (Cohen, Caparulo, Shaywitz, and Bower, 1977). In addition, the degree of elevation of HVA in urine in Figure 1 was correlated with the increased severity of autistic symptoms (Garreau et al., 1980).
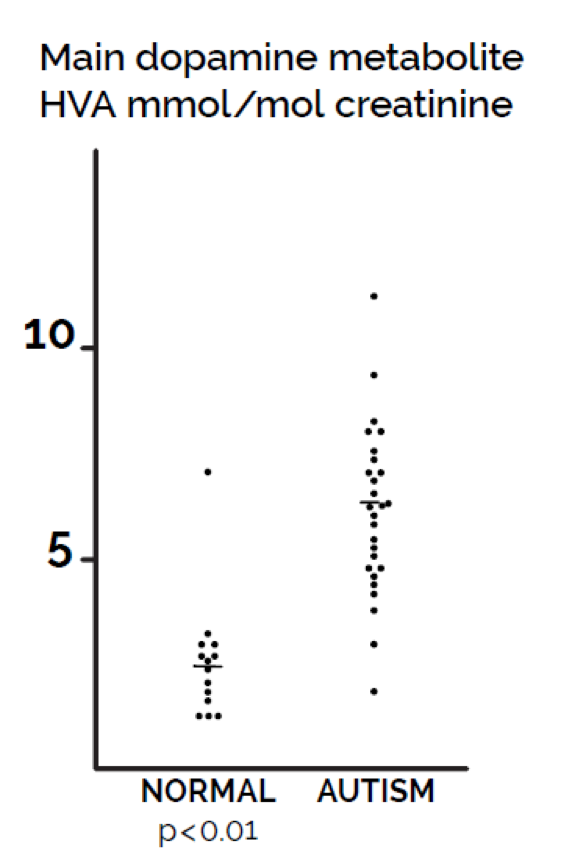
Figure 1. Comparison of values of urine dopamine metabolite homovanillic acid (HVA) in children with autism compared with controls of the same age range. Statistical significance (p< 0.01) based on t-test.
Thus, it seems very worthwhile to examine potential causes of excess dopamine in autism and the mechanisms by which excess dopamine alters behavior and causes brain damage. The purpose of the current review is to explore the genetic, toxic, and infectious factors that alter dopamine metabolism to show how all of these factors appear to cause autism by causing an excess of dopamine and a deficiency of norepinephrine and epinephrine in at least a substantial subgroup of people with ASDs.
The current article also adds new research and also reviews the evidence that helps to explain the role of excess dopamine in a previous study of an animal model of autism and its relevance to human autism. In addition, this focus on dopamine metabolism can also help to explain the immune system abnormalities common in autism. I also present new research and review previous research on the alteration of dopamine metabolism by microbial metabolites prevalent in urine samples of people with autism and a major environmental factor that has altered the microbiome to increase those species of bacteria, the metabolites of which alter dopamine metabolism.
The only drug currently approved by the USA Food and Drug Administration for the treatment of autism is risperidone (Risperdal), a drug designed to block dopamine receptors (Sharma and Shaw, 2012). It is an antagonist of the D1 (D1, and D5) as well as the D2 family (D2, D3 and D4) receptors, with 70-fold selectivity for the D2 family (Olguín, Guzmán, García, and Mejía, 2016). The drug previously used widely for autism treatment was haloperidol, also a dopamine blocking drug (“Risperdal: New indication”, 2006).
The metabolism of catecholamine neurotransmitters has been reviewed by Robertson et al (1991). The production of the catecholamine neurotransmitters in humans follows a similar pathway in the brain, the post-ganglionic neurons of the sympathetic nervous system, and the adrenal gland (Figure 2). In humans, phenylalanine and/or tyrosine from dietary proteins or amino acid supplements are absorbed from the intestinal tract where these amino acids cross the blood-brain barrier and enter the brain. Phenylalanine in the brain is converted to tyrosine by phenylalanine hydroxylase. The ring of tyrosine is then hydroxylated to dihydroxy-phenylalanine (DOPA) by tyrosine hydroxylase. DOPA is then converted to dopamine by DOPA decarboxylase which requires a vitamin B6 cofactor. The fate of further dopamine metabolism depends on the neuron type. In dopamine-containing neurons, the final product is dopamine. Dopamine, at normal concentrations in the neurons, is metabolized primarily to 3, 4-dihydroxyphenylacetic acid (DOPAC) and then to homovanillic acid (HVA), both of which can be measured in the urine with a number of different methods.
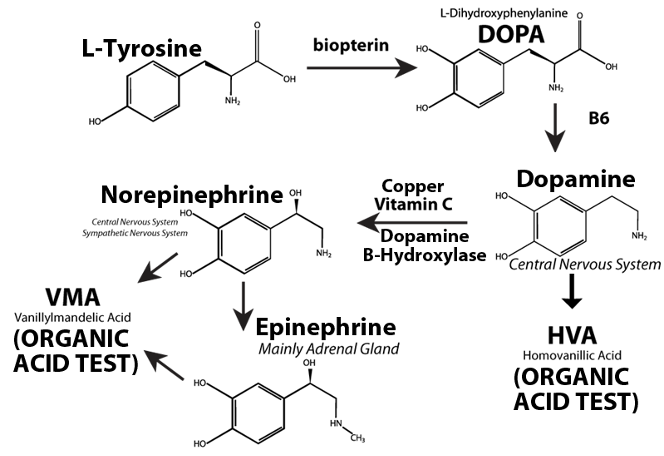
Figure 2. Biosynthesis of catecholamine neurotransmitters in the central and peripheral nervous system
In norepinephrine-containing brain neurons, neurons of the peripheral central nervous system, and the adrenal gland, dopamine is converted to norepinephrine by dopamine-beta-hydroxylase (DBH) present in synaptic vesicles. DBH requires ascorbic acid and copper as cofactors. In the adrenal gland, norepinephrine is further converted to epinephrine by phenylethanolamine N- methyl transferase. Both epinephrine and norepinephrine may then be converted to vanillylmandelic acid (VMA). DBH levels in cerebrospinal fluid correlate very highly with those in serum so that serum DBH allows one to indirectly assess DBH in the brain (Chen et al, 2010).
Damage to neurons due to excessive dopamine
Dopamine in the cytosol at pH 7.4 is unstable because the protons of the hydroxyl groups are dissociated and much more rapidly converted to unstable quinones that generate severe oxidative stress (Figure 3). Each molecule of unstable dopamine quinone generates an excess of oxygen superoxide radicals. In addition, cysteine and glutathione adducts of these unstable quinones are converted to metabolites that cause apoptosis of neurons. Dopamine uptake into monoaminergic vesicles prevents both the accumulation of free dopamine in the cytosol and the oxidation of dopamine to o-quinones and amino acid adducts because the pH inside monoaminergic synaptic vesicles is 2.0–2.4 pH units lower than the pH in the cytosol (Munoz, Huenchuguala, Paris, and Segura, 2012). The optimum enzyme activity of DBH corresponds to the low pH of the synaptic vesicles. At this low pH, the protons of dopamine hydroxyl groups are strongly bound to the oxygen of hydroxyl groups. When dopamine concentrations in the cytosol exceed the capacity of the synaptic vesicles to remove excess cytosol dopamine, the production of unstable quinones, toxic adducts of dopamine, and oxygen superoxide increases, leading to increasing oxidative stress, damage to neuronal mitochondria by covalent bonding of the dopamine metabolite aminochrome to the mitochondrial complexes I and III of the electron transport chain and to the Krebs cycle isocitrate dehydrogenase, neuron damage, and neuronal apoptosis (Figure 4) (Paris et al., 2011).
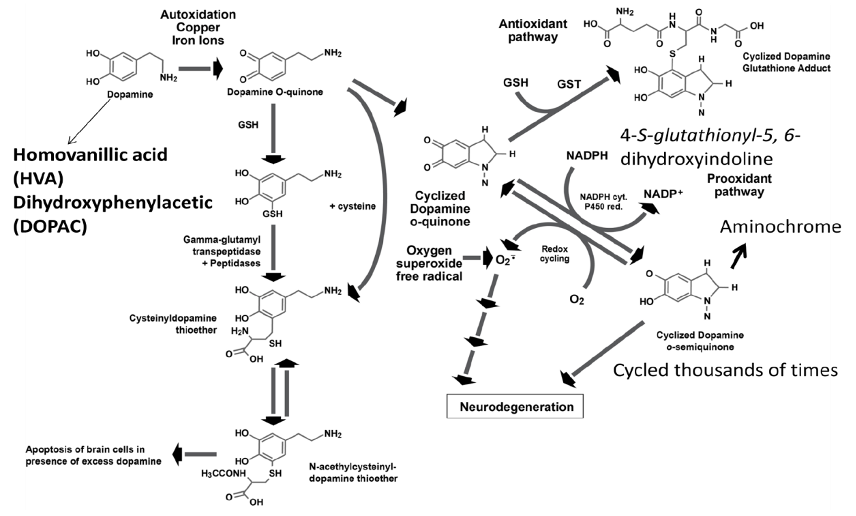
Figure 3. Formation of toxic dopamine quinone metabolites and oxygen superoxide free radicals when dopamine is elevated due to reduced dopamine beta-hydroxylase deficiency caused by excessive Clostridia enzyme inhibitors and/or genetic enzyme deficiency.
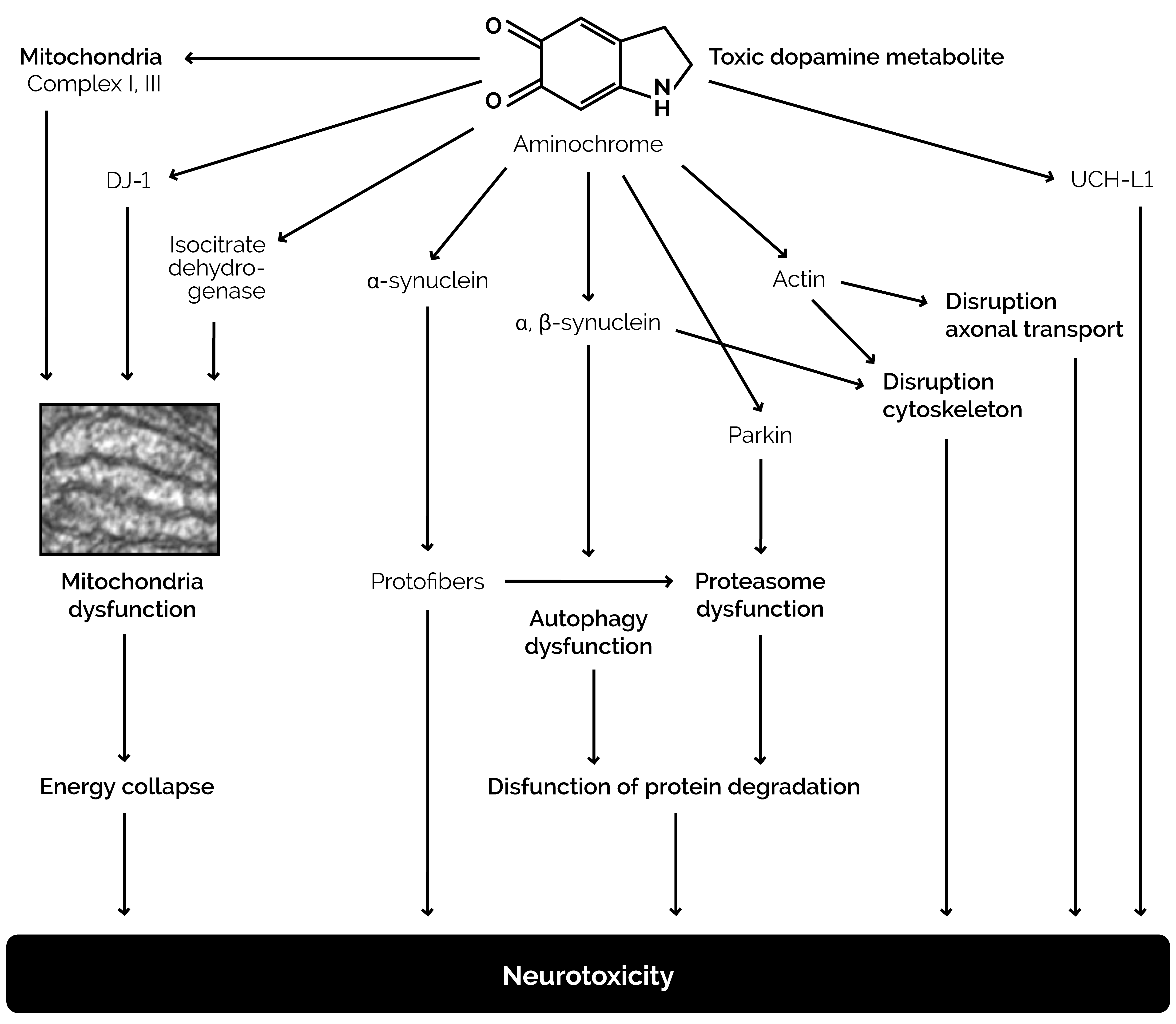
Figure 4. Damage to neurons caused by excessive dopamine metabolite aminochrome, the formation of which is indicated in Figure 3.
Properties and genetics of DBH
The functional DBH enzyme has four subunits. Each subunit is a 617 amino acid protein in length with 15 cysteine residues of which (Miyazaki and Asanuma, 2008) are involved in disulfide bond formation with a 25 amino-acid signal peptide that is removed at the time of secretion (Kapoor, Shandilya, and Kundu, 2011). The DBH gene has been mapped to chromosome 9 at the q34 position (Mustapic et al., 2014). Polymorphisms of the DBH gene have been identified in just 80 subjects: 68 single nucleotide polymorphisms (SNPs), 2 insertion/deletions and 1 repeat and 18 of the SNPs had minor allele frequencies greater than 5% Chen et al., 2010). The enzymatic activity of DBH in plasma corresponds to the level of DBH protein and is relatively stable over time in the same person, and family studies indicate that the effects of environmental factors such as stress and medications on DBH activity are small compared to genetic factors (Chen et al., 2010). The DBH insertion/deletion variant with a 19-bp sequence 118 bp downstream from the 5’-flanking region of the gene is the most common insertion/deletion variant. Individuals with 2 of the deletions have about half of the DBH activity as those with no deletions. Heterozygotes have an intermediate value. African Americans, European Americans, and Japanese populations all possess a common SNP 1021 C-T. Individuals with 2 of the T alleles have very low DBH activity while those with one T allele have intermediate enzyme activity and those with 2 of the C alleles have high enzyme activity. There are a very large number of possible combinations of
variants for DBH (4,970 combinations) based on the 71 genetic variants mentioned above but the amount of enzyme activity has been published for only the most common SNPs (Chen et al., 2010). Approximately 8% of a randomly selected population was found to have a thermolabile form of serum DBH that exhibited familial aggregation, indicating that SNPs of the enzyme are common (Robertson et al., 1991).
DBH and autism
Robinson et al (Robinson, Schutz, Macciardi, White, and Holden, 2001) found that the mean serum DBH was 30% lower in mothers of children with autism compared to mean values of those without a child with autism, indicating a likely genetic involvement of DBH as a cause of autism. In the same study, it was found that mothers of any sons with autism had a higher incidence (33%) of homozygous 19 base pair deletion polymorphism (DBH-) in the DBH gene compared to 20% of controls. The epidemiological term “attributable risk” for this gene was 42%, interpreted as meaning that the risk of having a son with autism was 42% higher for the mothers with this genetic variant than without it. DBH genotypes also differed significantly among mothers and controls in those with two sons with autism, with 37% of mothers with two affected sons having two DBH− alleles, compared to 19% of controls. The DBH− allele was associated with lower mean serum DBH enzyme activity in a pooled sample of mothers and controls. These researchers suggest that lowered maternal serum DBH activity results in a suboptimal uterine environment (decreased norepinephrine relative to dopamine), which, in conjunction with genotypic susceptibility of the fetus, results in autism spectrum disorder in some families. Unfortunately, the children with autism of these mothers with low DBH were not tested for low DBH. However, a high incidence of hyperflexible joints and postural hypotension has been reported in autism, implicating a potential role of DBH deficiency as a cause of autism.
Thomas et al (Thomas, Matsumoto, and Palmiter, 1995) used gene targeting to produce mice that lack DBH and are therefore unable to synthesize norepinephrine and epinephrine. They found that in heterozygous mothers, most homozygous embryos died in utero and only about 5% reached adulthood. Survival probably depended on catecholamine transfer across the placenta, because in homozygous deficient mothers all embryos died in utero. Mortality was due to lack of norepinephrine in utero because it could be prevented by treatment with the drug dihydroxyphenylserine (DOPS), a precursor of norepinephrine that can be converted to norepinephrine in the absence of DBH.
DBH deficiency also had a profound effect on maternal behavior of mice. Thomas and Palmiter (1997) found impaired maternal behavior in these mice with targeted disruption of the DBH gene. Most heterozygous pups born to DBH -/-females died within several days of birth and were often found scattered within the bedding. Potential causes, including deficits in olfaction and lactation, were not apparent. A deficit in maternal behavior was confirmed by the lack of pup retrieval exhibited by DBH -/- virgin females. Restoration of norepinephrine shortly before but not after birth induced females that had previously abandoned their litters to act maternally. These results suggested to the authors that norepinephrine is responsible for long-lasting changes that promote maternal behavior during both development and parturition in mice.
In a mouse model of autism Hsiao et al (2013) found that a metabolite of ethylphenol which is a Clostridia bacteria metabolite (Smith and MacFarlane, 1997) and a potent inhibitor of DBH (Goodheart, DeWolf, Jr., and Kruse, 1997) induced a severe anxiety state similar to autism in mice and that oral treatment of affected mice with the human commensal Bacteroides fragilis corrects gut permeability, alters microbial composition, and ameliorates defects in communicative, stereotypic, anxiety-like, and sensorimotor behaviors.
Metabolites of Clostridia species and their effect on DBH
Metabolites of certain Clostridia bacteria species including 4-cresol and 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA) acid are inhibitors of DBH and elevated levels of these compounds are prevalent in autism (Shaw, 2010). Shaw first identified HPHPA in urine and reported increases of HPHPA in 2010 in urine of children with autism and schizophrenia but also in most other neuropsychiatric diseases as well (Shaw, 2010); he also demonstrated that symptoms of schizophrenia disappeared after treatment with antibiotics that kill Clostridia without the use of neuroleptic drugs. Since then, the finding of high HPHPA values in autism has been confirmed by research groups in China (Xiong, Liu, Wang, Zeng, and Peng 2016) Turkey (Keşli , Gökçen, Buluğ, and Terzi, 2014), and Italy (Noto et al., 2014). The mechanism of DBH inhibition (Figure 5) has been documented especially well for 4-cresol (Southan, DeWolf Jr, and Kruse, 1990).
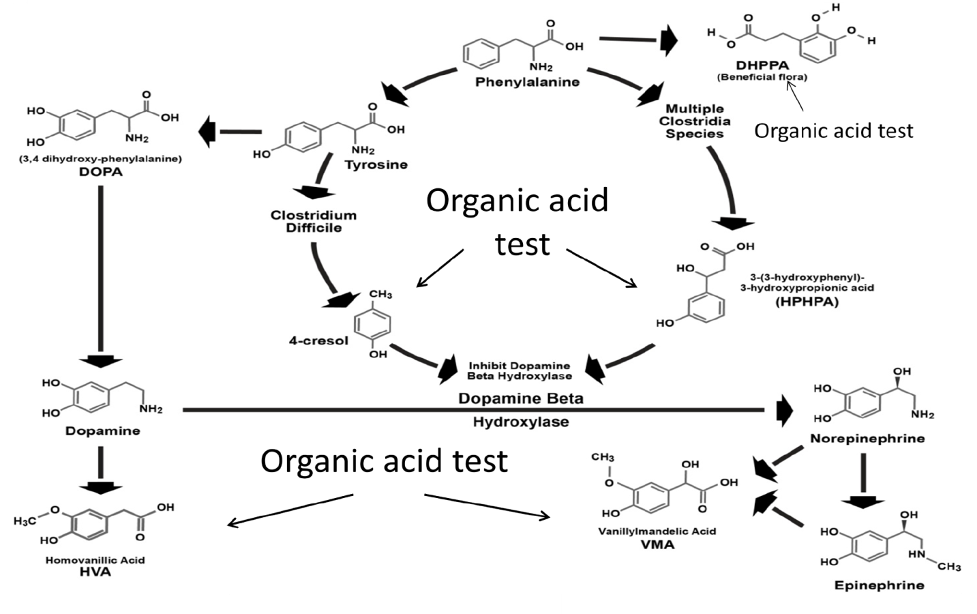
Figure 5. Combined human and Clostridia metabolism showing inhibition of dopamine beta-hydroxylase by Clostridia metabolites 4-cresol and 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA) and compounds in the pathway measured by organic acid testing.
4-Cresol forms covalent bonds with two different tyrosine positions (amino acids 216 and 357) of the DBH enzyme, which cause irreversible inhibition of the DBH enzyme activity (Southan et al., 1990; DeWolf et al., 1988) Thus, the presence of high amounts of the Clostridia metabolites may result in a reduction in DBH similar to genetic deficiency of DBH, resulting in dopamine excess and norepinephrine and epinephrine deficiencies. Indeed, Shaw has reported (Shaw, 2015) a wide variety of neurological and psychiatric diseases associated with high amounts of these Clostridia markers in the urine. Since homovanillic acid (HVA) is the major metabolite of dopamine and vanillylmandelic acid (VMA) is the major metabolite of both epinephrine and norepinephrine, the measurement of the HVA/VMA ratio is a good indicator of inhibition of DBH in the presence of high Clostridia metabolites.
Since DBH activity is inhibited by Clostridia, the variability in the enzyme activity of DBH might be much higher due to the genetic differences in the effects of Clostridia metabolites on DBH enzymes with different primary amino acid sequences due to genetic polymorphisms. Figure 6 demonstrates the correlation between the urinary excretion of the Clostridia metabolite HPHPA and the dopamine metabolite HVA in urine samples of a child with especially severe autism over 4 years (Shaw, 2017).
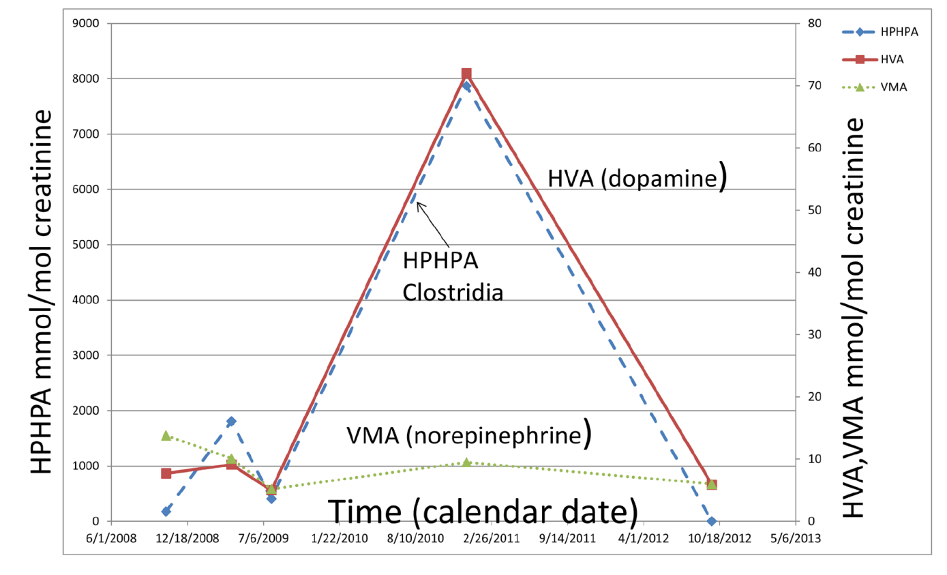
Figure 6. Relationships among Clostridia metabolite 3-(3-hydroxyphenyl) 3-hydroxypropionic acid (HPHPA), a major dopamine metabolite, homovanillic acid (HVA), and a major norepinephrine and epinephrine metabolite vanillylmandelic acid (VMA) over a period of 4 years in urine samples of a child with severe autism. Note the almost perfect correspondence between HPHPA and HVA.
The degree of elevation of HVA exactly parallels that of HPHPA for the entire time period, consistent with HPHPA inhibition of DBH. Meanwhile the concentration VMA, the major metabolite of norepinephrine and epinephrine is much lower the entire time period, again reflecting the severe inhibition of DBH. The amount of the dopamine metabolite HVA at the peak concentration, 72mmol/mol creatinine, is 5.53 times the upper limit of the age-related normal range and is perhaps the highest urinary HVA ever reported for a child in the medical literature, except for patients with neuroblastoma or pheochromacytoma, diseases in which the concentrations of HVA and/or VMA in the urine are commonly very high due to the catecholamine-secreting tumors. At the peak HVA concentration, the concentration of HPHPA, 7873 mmol/mol creatinine, was also extremely high, 35.8 times the upper limit of the sex and age-related normal range of the author’s laboratory.
Glyphosate and the microbiome
Glyphosate is one of the most prevalent chemicals used in agriculture. Some countries use glyphosate in greater amounts than all other agricultural chemicals combined. Monsanto brought it to market in 1974 under the trade name RoundupTM, and Monsanto’s last commercially relevant United States patent expired in 2000 (Shaw, 2015) Many companies now produce the herbicide. By 2016 there was a 100-fold increase from the late 1970s in the frequency of application and volume of glyphosate-based herbicides applied, with further increases expected in the future, partly in response to the global emergence and spread of glyphosate-resistant weeds (Shaw, 2015). The shikimic acid pathway of weeds and common non-genetically modified (non-GMO) food plants produces aromatic amino acids needed for plant growth; glyphosate inhibits a key enzyme in this pathway, leading to the death of the weed or non-GMO plant.
Although glyphosate was designed to kill weeds, it also kills susceptible bacteria that have biochemical pathways similar to plants and weeds. Shehata et al (Shehata, Schrödl, Aldin, Hafez, Krüger, 2013) found that glyphosate exposure in poultry can lead to a marked increase in pathogenic bacteria, such as Salmonella and Clostridia species, that were resistant to glyphosate in the stool samples of the poultry and to a significant decrease in beneficial flora, such as Enterococcus faecalis, Enterococcus faecium, Bacillus badius, Bifidobacterium adolescentis, and Lactobacillus species, which are susceptible to glyphosate.
In addition, ingestion of the herbicide could be a significant predisposing factor that has been associated with an increase in diseases mediated by Clostridium botulinum (C botulinum) (Krüger, Shehata, Schrödl, and Rodloff, 2013; Rulff, Schrödl, Basiouni, Neuhaus, and Krüger, 2015) in cattle, and perhaps, even in the farmers exposed to infected cattle (Rodloff and Kruger, 2012). C. botulinum is one of 3 species of Clostridia bacteria that produces large quantities of the precursors of 3-[3-hydroxyphenyl]-3- hydroxypropionic acid (HPHPA), a phenolic compound (Shaw, 2015). Microorganisms in the soil employ the shikimate pathway similar to plants to synthesize their aromatic amino acids so the composition of the soil bacteria in fields sprayed with glyphosate is altered as well. (Aristilde et al., 2017)
Shaw reported (2017) on a case study of triplets exposed to high amounts of glyphosate due to excessive ingestion of GMO corn tortillas. Two of the children had autism while a third had a suspected seizure disorder. All of the children had high biochemical markers indicating mitochondrial damage. All three children had markedly elevated urinary glyphosate with their mean baseline value—34.4 μg/g creatinine—being 25.5 times the median value of 1.35 μg/g creatinine and 24.1 times the mean value of 1.43 μg/g creatinine of the current study’s internal reference range. The glyphosate value in urine in one of the retested triplets decreased dramatically (94% decrease) after switching to an organic food diet. Symptoms of autism declined considerably after organic diet implementation.
All of the triplets had elevated succinic acid in their urine, an indicator of mitochondrial dysfunction (Shaw), on at least one of the two organic acids tests performed on these patients. Both males had elevated succinic acid on one of the two samples, whereas the female had elevated values on both organic acids tests.
Succinic acid is metabolized by the enzyme succinic dehydrogenase, which is significant in that it is both a Krebs cycle enzyme and a component— complex 2—of the mitochondrial electron transport chain, making this metabolite a marker of mitochondrial complex 2 dysfunction. Succinic acid has been found to be a sensitive marker for a variety of other toxic chemicals as well, including both metal and nonmetal toxic chemicals.
In addition to elevated markers associated with mitochondria damage, a high quantity of phenolic compounds produced by Clostridia bacteria in the two male members of the triplets is consistent with the previous reports that glyphosate can alter the intestinal flora of exposed animals and human. Both the males with the elevated Clostridia markers had depressed conversion of dopamine to norepinephrine, leading to overproduction of dopamine, a feature prevalent in autism (Shaw, 2017).
This case study which provides a plausible biochemical mechanism connecting glyphosate and autism is very important considering the epidemiological evidence that shows a strong connection between glyphosate usage and the increased autism incidence (Swanson, Leu, Abrahamson, and Wallet, 2014) (Figure 7). Autism was present at very low rate in the early 1990s but began significant increases by the late 1990s along with the increased production of genetically modified corn and soy modified to be glyphosate resistant.
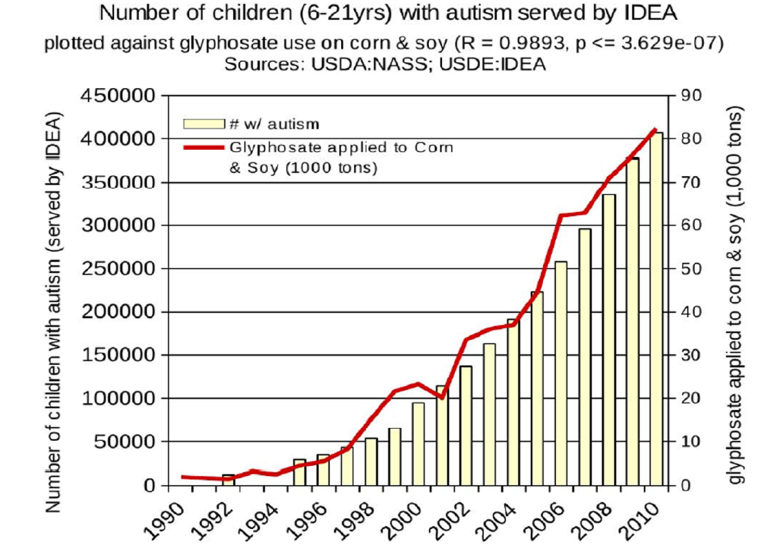
Figure 7. Correspondence between the agricultural use of glyphosate on corn and soy crops and the incidence of autism.
Immune system abnormalities, DBH deficiency, and autism
A large number of immune disorders have been documented in autism including low numbers of natural killer cells, increased T-cell abnormalities, IgG deficiency, IgG subclass deficiencies, IgA deficiency, increased IgE, increased antibodies to myelin basic proteins, increased amounts of IgG antibodies to wheat and milk proteins, defective immune response to vaccination, complement component deficiencies, increased family history of autoimmune disease (Edmiston, Ashwood, and Van de Water, 2016), and increased amounts of maternal antibodies against brain proteins. What is the effect of DBH deficiency associated with increased dopamine and reduced epinephrine and norepinephrine on the immune system?
Norepinephrine, released from sympathetic neurons, and epinephrine, released from the adrenal medulla, participate in a number of physiological processes including those that facilitate adaptation to stressful conditions. The thymus, spleen, and lymph nodes are richly innervated by the sympathetic nervous system, and catecholamines are thought to modulate the immune response (Alaniz et al., 1999). Alaniz and his group reported that 32% of DBH deficient mice which are deficient in epinephrine and norepinephrine died during adolescence but that these premature deaths did not occur when such mice were housed in a specific pathogen-free environment, suggesting that these mice may be more susceptible to infection (Alaniz et al., 1999). When housed in specific pathogen-free conditions, and in the absence of an infectious challenge, the immunological status of these mice appeared to be normal. However, when they were infected with Listeria monocytogenes or Mycobacterium tuberculosis, or were immunized with a T cell-dependent antigen, the mice were more susceptible to infection, had impaired T cell function, and had impaired Th1 T cell-dependent-IgG2a antibody production. The DBH deficient mice also produced markedly increased corticosteroids after infection which may have suppressed immune response.
Pharmaceutical treatment of DBH deficiency to prevent autism
The lack of norepinephrine and epinephrine due to deficiency of DBH due to genetic reasons cannot be resolved by supplementation of phenylalanine, tyrosine, or L-DOPA since these agents lead to overproduction of dopamine and its toxic metabolites before adequate amounts of norepinephrine and epinephrine can be formed. The formation of these toxic metabolites of dopamine would presumably be accelerated if there are SNPs leading to low activities of catechol-O-methyltransferase (COMT) or monoamine oxidase (MAO) or deficiencies of their cofactors like S-adenosyl methionine. The drug L-threo-dihydroxyphenylserine can be converted directly to norepinephrine by the action of amino acid decarboxylase (Chen et al., 2010). L-threo-dihydroxyphenylserine has been very effective in the treatment of genetic DBH deficiency (Chen et al., 2010) and has also been used for the treatment of Parkinson’s disease (Hauser, Biaggioni, Hewitt, and Vernino, 2018). Although not evaluated for the prevention of autism in women with DBH deficiency or treatment of autism in children with genetic DBH deficiency, a clinical trial of the drug to treat DBH deficient mothers and children in order to prevent the development of autism would seem to be worthwhile after more extensive animal experiments are conducted to establish safety.
Summary
In summary, for forty years abnormally elevated dopamine has been noted in the body fluids of people with autism and was treated with dopamine receptor blocking drugs like risperidone and haloperidol. Elevated dopamine and reduced norepinephrine and epinephrine may be caused by genetic deficiency of DBH in utero and/or postnatally or by inhibition of DBH by a number of phenolic compounds produced by Clostridia bacteria species. Such Clostridia bacteria and their phenolic metabolites are prevalent in autism and are potent DBH inhibitors. Glyphosate, one of the most widely used herbicides worldwide, is strongly correlated in time with the increased incidence of autism. The use of this herbicide has caused increased incidence of farm animals colonized or infected with Clostridia species that are resistant to the antibacterial properties of glyphosate. The control of these Clostridia species may be essential to the treatment of many people with autism. The most effective way to reduce the presence of such Clostridia species would appear to be limiting the use of glyphosate in agriculture. Overcoming the effects of DBH deficiency with drugs that provide norepinephrine by an alternative biochemical pathway is a potential new therapy for those people with autism with genetic DBH deficiency that should be explored. An overall summary of these factors is given in Figure 8.
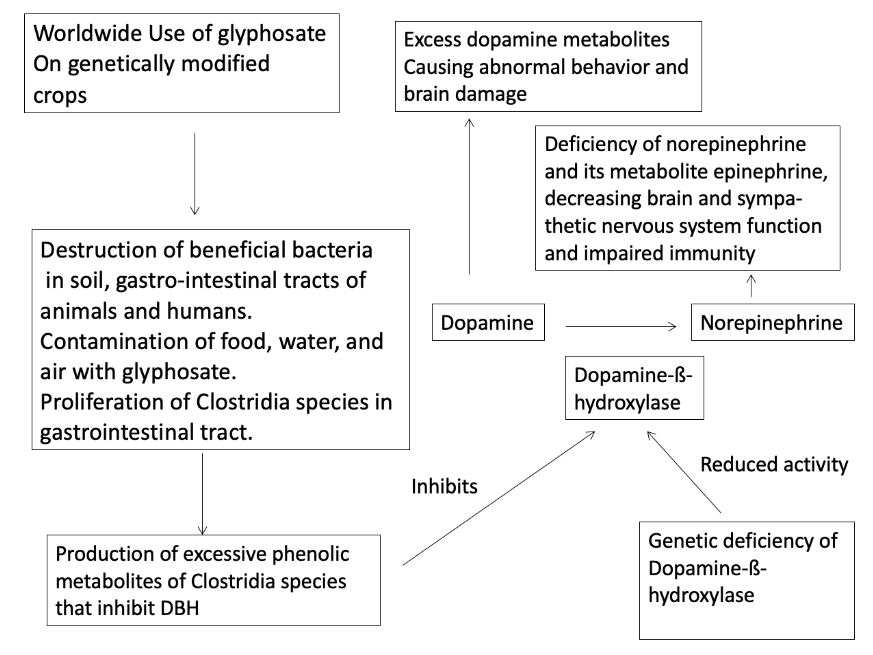
Figure 8. Summary of proposed mechanism of autism epidemic due to environmental, microbial, and genetic factors.
References
Alaniz RC, Thomas SA, Perez-Melgosa M, Mueller K, Farr AG, Palmiter RD & Wilson CB (1999) Dopamine b-hydroxylase deficiency impairs cellular immunity. Proceedings of the National Academy of Sciences of the United States of America. USA. 96: 2274–2278
Aristilde L, Reed ML, Wilkes RA, Youngster T, Kukurugya MA, Katz V & Sasaki C (2017) Glyphosate-induced specific and widespread perturbations in the metabolome of soil Pseudomonas species. Frontiers in Environmental Science. 5:34. doi: 10.3389/fenvs.2017.00034
Chen Y, Wen G, Rao F, Zhang K, Wang L, Rodriguez-Flores JL & O’Connor DT (2010) Human dopamine beta-hydroxylase (DBH) regulatory polymorphism that influences enzymatic activity, autonomic function, and blood pressure. Journal of Hypertension. January; 28(1): 76–86. doi:10.1097/HJH.0b013e328332bc87
Cohen DJ, Caparulo BK, Shaywitz BA & Bower MB Jr. (1977) Dopamine and serotonin metabolism in neuropsychiatrically disturbed children: CSF homovanillic acid and 5-hydroxyindoleacetic acid. Archives of General Psychiatry. 34: 545-550.
DeWolf WE Jr., Carr SA, Varrichio A, Goodhart PJ, Mentzer MA, Roberts GD & Kruse LI (1998) Inactivation of dopamine beta-hydroxylase by p-cresol: Isolation and characterization of covalently modified active site peptides. Biochemistry. 27 (26):9093-9101.
Edmiston E, Ashwood P & Van de Water J (2017) Autoimmunity, autoantibodies, and autism spectrum disorders (asd). Biological Psychiatry. 81(5): 383–390. doi:10.1016/j.biopsych.2016.08.031
Garreau B, Barthelemy C, Domenech J, Sauvage D, Muh JP, Lelord G & Callaway E (1980) Disturbances in dopamine metabolism in autistic children: results of clinical tests and urinary dosages of homovanilic acid (HVA). Acta Psychiatrica Belgica. 80(3):249-265.
Goodhart PJ, DeWolf WE Jr. & Kruse LI (1987) Mechanism-based inactivation of dopamine β-hydroxylase by p-cresol and related alkylphenols. Biochemistry. 26: 2516–2583.
Hauser RA, Biaggioni I, Hewitt LA & Vernino S (2018) Integrated analysis of droxidopa for the treatment of neurogenic orthostatic hypotension in patients with parkinson disease. Movement Disorders Clinical Practice. 5(6):627-634. doi: 10.1002/mdc3.12695.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T & Masmanian SK (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 155(7):1451-1463.
Kapoor A, Shandilya M & Kundu S (2011) Structural insight of dopamine b-hydroxylase, a drug target for complex traits, and functional significance of exonic single nucleotide polymorphisms. PLoS ONE. 6(10): e26509.
Keşli R, Gökçen C, Buluğ U & Terzi Y (2014) Investigation of the relation between anaerobic bacteria genus Clostridium and late onset autism etiology in children. Journal of Immunoassay and Immunochemistry. 35(1):101-109.
Krüger M, Shehata AA, Schrödl W & Rodloff A (2013) Glyphosate suppresses the antagonistic effect of Enterococcus spp. on Clostridium botulinum. 20:74-78. doi: 10.1016/j.anaerobe.2013.01.005. EPub. Feb 6.
Lelord G, Callaway E, Muh JP, Arlot JC, Sauvage D, Garreau B & Domanech J (1978) Modifications in urinary homovanillic acid after ingestion of vitamin B6; functional study in autistic children. Revista de Neurologia. 134 (12):797-801.
Miyazaki I & Asanuma M (2008) Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Medica Okayama. 62(3):141-150.
Munoz P, Huenchuguala S, Paris I & Segura-Aguilar J (2012) Dopamine oxidation and autophagy. Parkinson’s Disease. 920953. doi:10.1155/2012/920953.
Mustapic M, Maihofer AX, Mahata M, Chen M, Baker DG, O’Connor DT & Nievergelt CM (2014) The catecholamine biosynthetic enzyme dopamine b-hydroxylase (DBH): First genome-wide search positions trait-determining variants acting additively in the proximal promoter. Human Molecular Genetics. 23: 6375–6384.
Noto A, Fanos V, Barberini L, Grapov D, Fattuoni C, Zaffanello M, Francavilla R (2014) The urinary metabolomics profile of an Italian autistic children population and their unaffected siblings. The Journal of Maternal-Fetal & Neonatal Medicine. 27 (sup2): 46- 52. DOI: 3109/14767058.2014.954784
Olguín HJ, Guzmán DC, García EH & Mejía GB (2016) The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxidative Medicine and Cellular Longevity. 9730467. http://dx.doi.org/10.1155/2016/9730467
Paris I, Munoz P, Huenchuguala S, Couve E, Sanders LH, Greenamyre JT & Segura-Aguilar J(2011) Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicological Sciences. 121(2): 376–388. doi:10.1093/toxsci/kfr060.
Pratt-Hyatt M, Haeusler RA, Yuen KC & Shaw W (2018) A single nucleotide polymorphism in the ARHGEF6 gene is associated with increased risk for autism spectrum disorder. Hereditary Genetics. 7:2 DOI: 10.4172/2161-1041.1000196.
Previc F (2007) Prenatal influences on brain dopamine and their relevance to the rising incidence of autism. Medical Hypotheses. 68: 46–60.
(2006). Risperidone: New indication. Behavioural disorders in children with autism or mental disabilities: No progress. Prescrire International. 82:43-45.
Robertson D, Haile V, Perry SE, Robertson RM, Phillips III JA & Biaggioni I (1991) Dopamine -beta-hydroxylase deficiency: A genetic disorder of cardiovascular regulation. Hypertension. 18: 1-8.
Robinson PD, Schutz CK, Macciardi F, White BN & Holden JJ (2001) Genetically determined low maternal serum dopamine beta-hydroxylase levels and the etiology of autism spectrum disorders. American Journal of Medical Genetics. 100(1):30-36.
Rodloff AC & Kruger M (2012) Chronic Clostridium botulinum infections in farmers. Anaerobe. 18(2):226-228.
Rulff R, Schrödl W, Basiouni S, Neuhaus J & Krüger M (2015) Is downer cow syndrome related to chronic botulism? Polish Journal of Veterinary Sciences. 18(4):759-765. doi: 10.1515/pjvs-2015-0098.
Sharma A & Shaw SR (2012) Efficacy of risperidone in managing maladaptive behaviors for children with autistic spectrum disorder: A meta-analysis. Journal of Pediatric Health Care. 26(4):291-299.
Shaw W (2010) Increased urinary excretion of a 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutritional Neuroscience. 13(3):135-43.
Shaw W (2015) Clostridia bacteria in the gastrointestinal tract as a major cause of depression and other neuropsychiatric disorders. Role of Clostridia metabolites 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA) and 4-cresol in the alteration of dopamine and norepinephrine metabolism. In: Greenblatt J, Brogan K, editors. Integrative psychiatry for depression: Redefining models for assessment, treatment, and prevention of mood disorders. New York: Taylor and Francis. p. 31-48.
Shaw W (2015) Exposure to toxic chemicals as a cause of depression. In: Greenblatt J, Brogan K, editors. Integrative psychiatry for depression: Redefining models for assessment, treatment, and prevention of mood disorders. New York: Taylor and Francis. p. 209-222.
Shaw W (2017) Elevated urinary glyphosate and Clostridia metabolites with altered dopamine metabolism in triplets with autistic spectrum disorder or suspected seizure disorder: A case study. Integrative Medice: A Clinician’s Journal (Encinitas). 16 (1):50-57.
Shaw W, Kassen E & Chaves E (1995) Increased urinary excretion of analogs of Krebs cycle metabolites and arabinose in two brothers with autistic features. Chemistry. 41(8): 1094-1104.
Shehata AA, Schrödl W, Aldin AA, Hafez HM & Krüger M (2013) The effect of glyphosate on potential pathogens and beneficial members of poultry microbiota in vitro. Current Microbiology. 66(4):350-358.
Smith EA & Macfarlane GT (1997) Formation of phenolic and indolic compounds by anaerobic bacteria in the human large intestine. Microbial Ecology. 33: 180–188.
Southan C, DeWolf WE Jr. & Kruse LI (1990) Inactivation of dopamine beta-hydroxylase by p-cresol: Evidence for a second, minor site of covalent modification at tyrosine 357. Biochimica et Biophysica Acta. 1037(2):256-258.
Swanson NL, Leu A, Abrahamson J & Wallet B (2014) Genetically engineered crops, glyphosate and the deterioration of health in the United States of America. Journal of Organic Systems. 9(2): 6-37
Thomas SA, Matsumoto AM & Palmiter RD (1995) Noradrenaline is essential for mouse fetal development. Nature. 374: 643-646.
Thomas SA & Palmiter RD (1997) Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell. 91: 583-592.
Xiong X, Liu D, Wang Y, Zeng T & Peng Y (2016) Urinary 3-(3-hydroxyphenyl)-3-hydroxypropionic acid, 3-hydroxyphenylacetic acid, and 3-hydroxyhippuric acid are elevated in children with autism spectrum disorders. BioMed Research International. http://dx.doi.org/10.1155/2016/9485412